Table Of Content
- Computational Design of Novel Enzymes Without Cofactors
- We're located in the Shriram Center for Bioengineering at Stanford,sharing a lab space together with the Bintu Lab.
- Design for affinity
- Symmetric Protein Architecture in Protein Design: Top-Down Symmetric Deconstruction
- Diffusion model expands RoseTTAFold’s power
- Data availability
- Generative models for protein structures and sequences

Through the years, our basic understanding of proteins has increased greatly, and we have begun to enter the era when we will be able to produce functional proteins that will revolutionize medicine and technology. Many of the hardest-to-treat diseases, such as Alzheimer's, many forms of cancer (e.g., TP53), and human immunodeficiency virus (HIV) infection involve protein–protein interactions. Thus, to treat such diseases, it is desirable to design protein or protein-like therapeutics that bind one of the partners of the interaction and, thus, disrupt the disease-causing interaction.
Computational Design of Novel Enzymes Without Cofactors
By varying the length of cODC1 and its attachment point to LOV2, Renicke et al. were ultimately able to isolate a module that rapidly and extensively reduced protein target concentrations upon illumination. Baker et al. designed and experimentally validated the first protein fold not found in nature, “Top7,” using their Rosetta-Design software.16,17 Their strategy was to construct the protein scaffold using three- and nine-residue fragments taken from the Protein Data Bank (PDB). The best combinations were then selected via Monte Carlo optimization of a number of energetic terms including hydrophobic burial, β-strand hydrogen bonding, and side-chain rotamers (Figure 3 right).
We're located in the Shriram Center for Bioengineering at Stanford,sharing a lab space together with the Bintu Lab.
Electron microscopy confirmed that the structures of these oligomers are very similar to the design models, which in many cases show little global similarity to known protein oligomers. A recent preprint from researchers at Generate describes a new generative modeling-based design algorithm called Chroma, which includes several features that improve its performance and success rate. These include diffusion models, an approach used in many image-generation AI tools that makes it easier to manipulate complex, multidimensional data. Chroma also employs algorithmic techniques to assess long-range interactions between residues that are far apart on the protein’s amino acid backbone, but that may be essential for proper folding and function.
Design for affinity
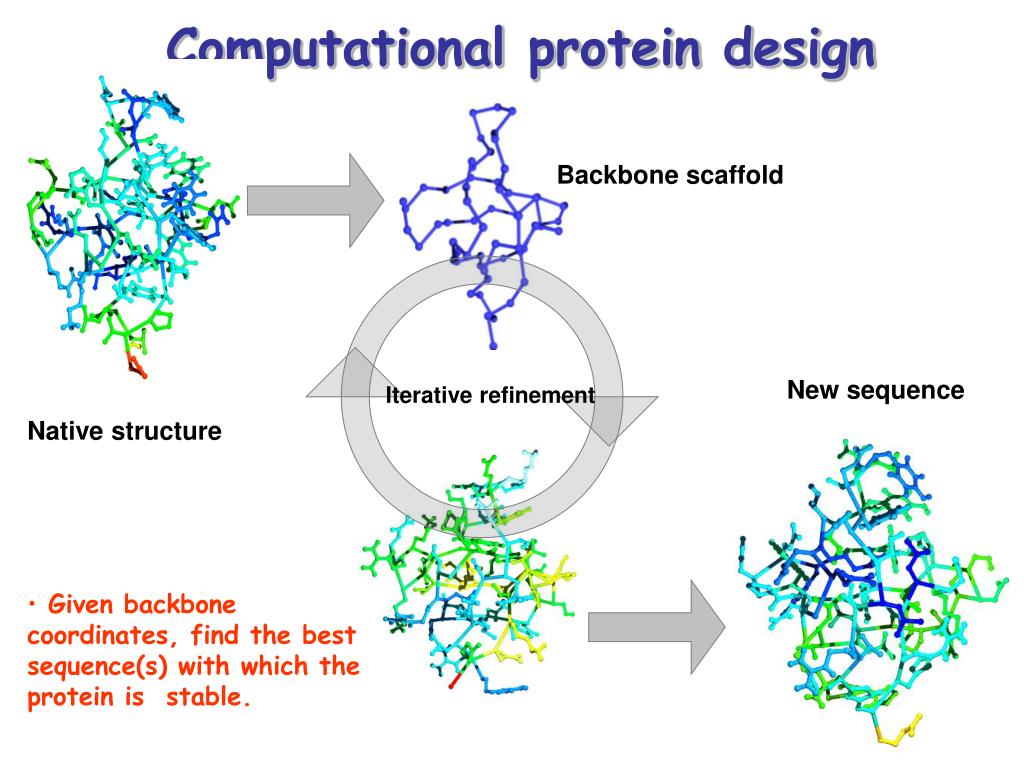
Following the publication of AlphaFold2 and RoseTTAFold in 2021, the field of protein structure prediction has moved quickly to incorporate these advances into protein engineering. Protein design (or protein engineering) is a technique by which proteins with enhanced or novel functional properties are created. Proteins can be engineered by rational design, which typically uses computational tools to identify useful mutations, or by directed evolution, which uses random mutagenesis coupled with a selection process to identify desired variants. The principles of physics play a crucial role in protein design, particularly in the development of computational models and simulations. By applying concepts from statistical mechanics, thermodynamics, and molecular dynamics, researchers can predict how a protein will fold and interact with other molecules in its environment.
“We’re starting to see the possibility of really truly creating a complex active site and then building the protein around it,” he says. For example, a protein with excellent catalytic properties might be exceedingly difficult to manufacture at scale or exhibit poor properties as a drug. In the future, however, next-generation algorithms should make it possible to generate de novo proteins optimized to tick off many boxes on a scientist’s wish list rather than just one. Some companies are also looking to augment public structural biology resources with data of their own. Generate is in the process of building a multi-instrument cryo-electron microscopy facility, which will allow them to generate near-atomic-resolution structures at relatively high throughput. Such internally generated structural data are more likely to include relevant metadata about individual proteins than data from publicly available resources.
Currently, most protein sequence design methods based on deep learning focus on network architecture optimization, while ignoring protein-specific physicochemical features. Inspired by the successful application of structure templates and pre-trained models in the protein structure prediction, we explored whether the representation of structural sequence profile can be used for protein sequence design. In this work, we propose SPDesign, a method for protein sequence design based on structural sequence profile using ultrafast shape recognition.
Data availability
However, through many such steps, the breadth of possible protein structures from which the input could have arisen narrows and RFdiffusion predictions come to closely resemble protein structures (Fig. 1c, right). We use the ProteinMPNN network1 to subsequently design sequences encoding these structures, typically sampling eight sequences per design in line with previous work5,16 (but see Supplementary Fig. 2a). We also considered simultaneously designing structure and sequence within RFdiffusion, but given the excellent performance of combining ProteinMPNN with the diffusion of structure alone, we did not extensively explore this possibility. Among the most challenging functions to design are conformational changes between multiple states. A single-state design would be successful as long as the designed state resides in a deep energy minimum, so that sizable scoring errors can often be tolerated (151). However, the multistate design (MSD) requires considerable accuracy in scoring relative stabilities, such that the probability distributions among multiple states can be modeled correctly.
De novo design of knotted tandem repeat proteins - Nature.com
De novo design of knotted tandem repeat proteins.
Posted: Tue, 24 Oct 2023 07:00:00 GMT [source]
Opportunities and challenges in design and optimization of protein function
A bold label design for a plant protein would likely involve bright, eye-catching colors and typography to capture consumers' attention. The design would also need to clearly communicate the benefits of the plant protein and appeal to consumers looking for healthy, sustainable protein options. The result would be a striking label design that stands out on the shelf and effectively communicates the product's value to consumers.
Generative models for protein structures and sequences
Advances in the design of protein switches that change conformation in response to diverse signals.A, a designed helical trimer changes its oligomerization state in response to pH changes (155). B, a designed helical bundle protein changes conformation upon binding to a calcium ion (green) and a chloride ion (blue) (156). C, a designed artificial chemically induced dimerization system (12) assembles upon binding to a farnesyl pyrophosphate ligand (spheres), linking ligand binding (sensing) to a modular response through reconstitution of a split output module (gray, magenta). D, in the LOCKR system, a helical peptide “key” (magenta) can displace and expose a signal peptide (green) (15). A recent method called AbDesign (66) seeks to mimic natural homologous recombination. In contrast to other methods, Abdesign uses larger segments and relies on the similarity between members of the same protein family to facilitate backbone sampling.
NsEM characterization of a C3 design (HE0822) with 350 residue subunits (1,050 residues in total) suggests that the actual structure is very close to the design, both over the 350 residue subunits and the overall C3 architecture. 2D class averages are clearly consistent with both top and side views of the design model, and a 3D reconstruction of the density has key features consistent with the design, including the distinctive pinwheel shape (Fig. 3b, top row). Electron microscopy 2D class averages of C5 and C6 designs with more than 750 residues (HE0794, HE0789, HE0841) were also consistent with the respective design models (Extended Data Fig. 5f). There is considerable interest in designing symmetric oligomers, which can serve as vaccine platforms34, delivery vehicles35 and catalysts36. Other strategies for de novo backbone generation do not use blueprints but still use assembly of protein fragments borrowed from nature.
In the simplest models, the protein backbone is kept rigid while some of the protein side-chains are allowed to change conformations. However, side-chains can have many degrees of freedom in their bond lengths, bond angles, and χ dihedral angles. To simplify this space, protein design methods use rotamer libraries that assume ideal values for bond lengths and bond angles, while restricting χ dihedral angles to a few frequently observed low-energy conformations termed rotamers. ML and other artificial intelligence (AI)-based computational tools have already proven their prowess at predicting real-world protein structures. AlphaFold 2, an algorithm developed by scientists at DeepMind that can confidently predict protein structure purely on the basis of an amino acid sequence, has become a household name since its launch in July 2021.







No comments:
Post a Comment